New
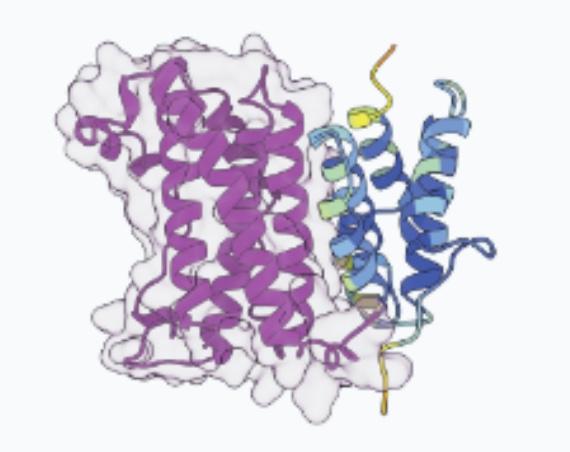
Orca-Fly: sequence-based 3D genome prediction in Drosophila
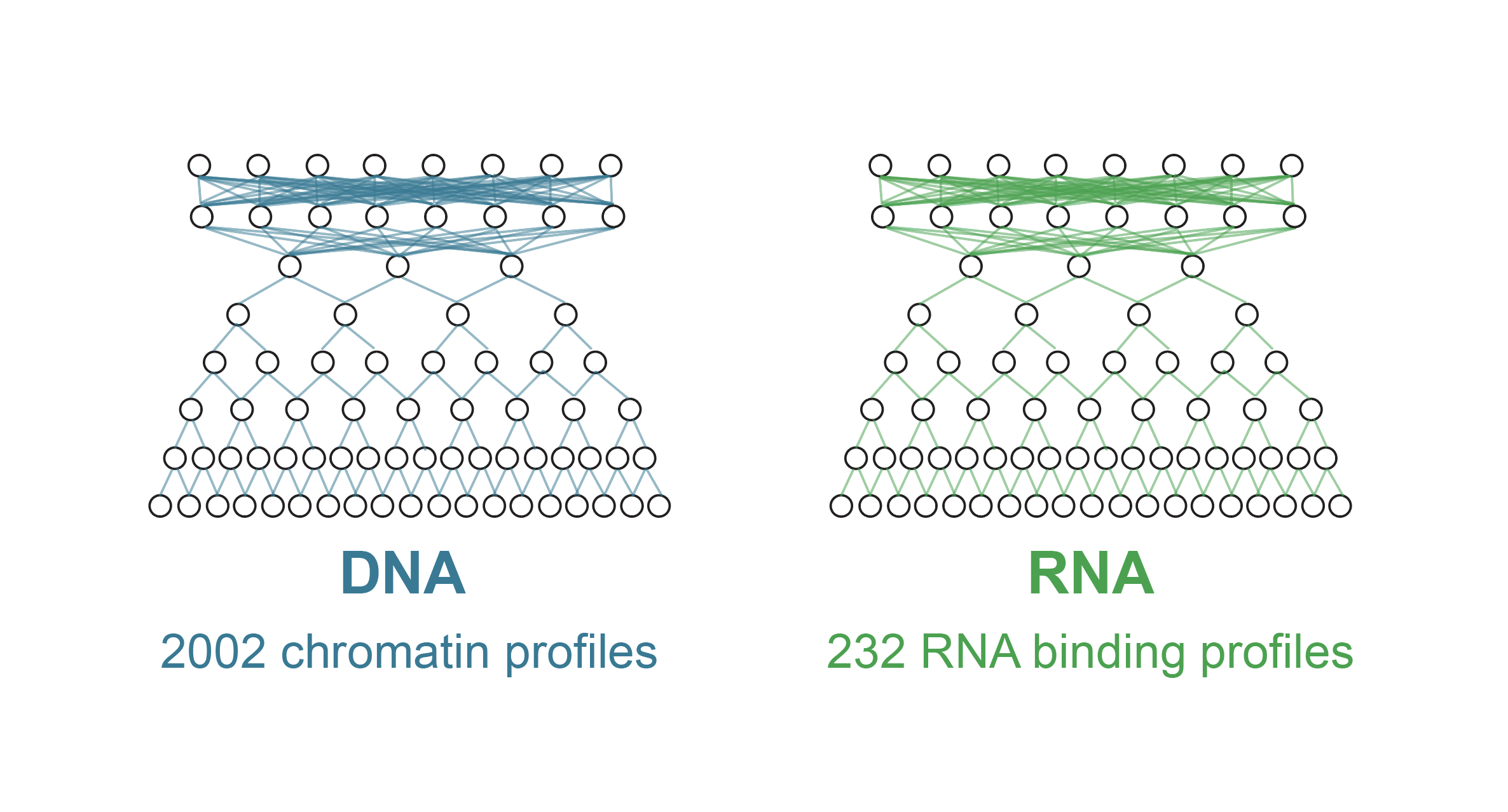
Orca-Fly extends the Orca framework to Drosophila, predicting multiscale 3D genome organization directly from genomic sequence. It enables sequence-based prediction and analysis of 3D genome architecture in the fly.